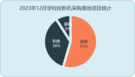
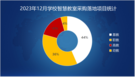
USEARCH算法在数据库中搜索对一个或多个数据库序列(“目标”)的高一致性命中。USEARCH由usearch_global和usearch_local命令使用,并由cluster_fast和cluister_smallmem用作子例程。该算法与为低身份本地搜索而设计的UBLAST算法从根本上不同。
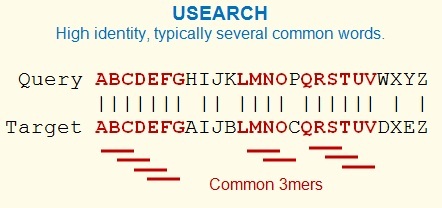
推荐的标识范围
USEARCH对蛋白质的识别率为50%及以上,对核苷酸的识别率为75%及以上时有效。
字数统计和U排序
USEARCH利用了这样的事实,即相似的序列往往有几个共同的短字。这些单词的长度固定为k,有时也称为kmers。与使用kmer计数的其他程序不同,USEARCH不会尝试根据匹配kmers的数量来估计序列同一性。这是因为身份仅与字数大致相关,而没有给出准确的估计,尤其是对于较低的身份。而是,USEARCH使用单词计数对数据库搜索进行优先级排序。按照减少唯一字数(U)的顺序将目标序列与查询进行比较。这是USEARCH中的“ U”。如果命中率超过了身份阈值 存在于数据库中,很可能在U排序目标列表(“ U向量”)的开头附近找到。
接受和拒绝
“接受”是高于同一性阈值的目标序列,因此可以视为命中。“拒绝”是与查询进行比较但低于阈值的目标序列。如果将目标与按U排序的查询进行比较,则:
(i)找到的第一个匹配可能是数据库中存在或接近数据库的最佳匹配;以及
(ii)没有命中发生的拒绝次数越多,存在命中的可能性就越小。
这意味着,如果(i)找到了一个命中点,或者(ii)发生了几次拒绝,则可以尽早终止搜索,而只损失很小的灵敏度。该终端选项 -maxaccepts和-maxrejects确定何时停止搜索。该技术可以显着提高速度,这对于生物学中越来越常见的非常大的序列数据集是决定性的优势。
单词索引
USEARCH使用数据库中单词的索引来快速计算给定查询的U向量。索引选项允许微调速度,灵敏度和内存使用。索引可以存储在UDB文件中,这在重复搜索大型数据库时可能是有利的。或者,例如,当以FASTA格式提供数据库时,可以动态构建索引。cluster_fast和cluister_smallmem也使用索引,它使用USEARCH作为子例程,以将查询序列与现有集群的质心数据库进行匹配。使用聚类时,由于必须在识别新的聚类时更新数据库,因此索引必须动态构建。